Analysis of reported adverse events of pipeline stents for intracranial aneurysms using the FDA MAUDE database
Article information

Abstract
Objective
Flow diverting stents (FDS) are a validated device in the treatment of intracranial aneurysms, allowing for minimally invasive intervention. However, after its approval for use in the United States in 2011, post-market surveillance of adverse events is limited. This study aims to address this critical knowledge gap by analyzing the FDA Manufacturer and User Facility Device Experience (MAUDE) database for patient and device related (PR and DR) reports of adverse events and malfunctions.
Methods
Using post-market surveillance data from the MAUDE database, PR and DR reports from January 2012-December 2021 were extracted, compiled, and analyzed with R-Studio version 2021.09.2. PR and DR reports with insufficient information were excluded. Raw information was organized, and further author generated classifications were created for both PR and DR reports.
Results
A total of 2203 PR and 4017 DR events were recorded. The most frequently reported PR adverse event categories were cerebrovascular (60%), death (11%), and neurological (8%). The most frequent PR adverse event reports were death (11%), thrombosis/thrombus (9%) cerebral infarction (8%), decreased therapeutic response (7%), stroke/cerebrovascular accident (6%), intracranial hemorrhage (5%), aneurysm (4%), occlusion (4%), headache (4%), neurological deficit/dysfunction (3%). The most frequent DR reports were activation/positioning/separation problems (52%), break (9%), device operates differently than expected (4%), difficult to open or close (4%), material deformation (3%), migration or expulsion of device (3%), detachment of device or device component (2%).
Conclusions
Post-market surveillance is important to guide patient counselling and identify adverse events and device problems that were not identified in initial trials. We present frequent reports of several types of cerebrovascular and neurological adverse events as well as the most common device shortcomings that should be explored by manufacturers and future studies. Although inherent limitations to the MAUDE database are present, our results highlight important PR and DR complications that can help optimize patient counseling and device development.
INTRODUCTION
Flow diverting stents (FDS) are a validated device in the treatment of intracranial aneurysms, allowing for minimally invasive intervention [28]. This stent allows blood to be diverted away from the aneurysm itself, thus taking pressure off the aneurysm to reduce risk of rupture by inducing aneurysm thrombosis and endothelialization of the device. FDS are often considered as a first choice of treatment for several aneurysms due to their high rate of successful management, low rate of aneurysm recanalization, ease of insertion, and short procedure duration [1,27].
Before the development of FDS, other endovascular procedures were available such as liquid embolic agents, balloon catheters, and intracranial stents to aid primary coil embolization in the treatment of cerebral aneurysms [24]. Despite these advancements, large (15-25 mm) and giant (>25 mm) wide-neck cerebral aneurysms remained technically challenging to treat, resulting in low elimination rates and high recurrence rates with endovascular treatment [4,8]. Thus, a novel solution to treat these types of aneurysms known as the Pipeline Embolization Device (PED) was granted FDA approval in 2011 for adult patients with wide-neck, large or giant aneurysms between the petrous and ophthalmic segments of the internal carotid artery [11]. Additionally, safe and effective off-label use of FDS has been reported for cerebral aneurysms in many locations with a parent vessel diameter of 2-5 mm [24].
Despite the large number of reports of successful use of FDS after its approval for use in the United States in 2011, post-market surveillance of adverse events is limited. The use of PEDs requires the use of dual-antiplatelet therapy to prevent periprocedural thromboembolic complications, which is associated with hemorrhagic complications [23]. This is an important consideration for long-term risk-benefit discussions when offering the various available treatment options. Post-market surveillance allows better identification of uncommon adverse events that may not have been appreciated in clinical trials due to smaller sample sizes and latency of events [15]. This study aims to address the critical knowledge gap of post-marketing surveillance and long-term safety monitoring by characterizing reported adverse events and device problems in patients treated with PEDs by querying data within the federal Manufacturer and User Facility Device Experience (MAUDE) database.
MATERIALS AND METHODS
Manufacturer and user facility device experience database
This study analyzed post-market surveillance data within the FDA MAUDE database. This is a publicly available database maintained by the FDA for tracking adverse events associated with medical devices approved for use in the United States [10,12]. Reporting to the database is required by manufacturers, importers, and device-user facilities. Reporting is voluntary by health care professionals, patients, and consumers. Since this database is publicly accessible and consists of deidentified data, no ethics approval was required.
Data mining, classification, and reporters
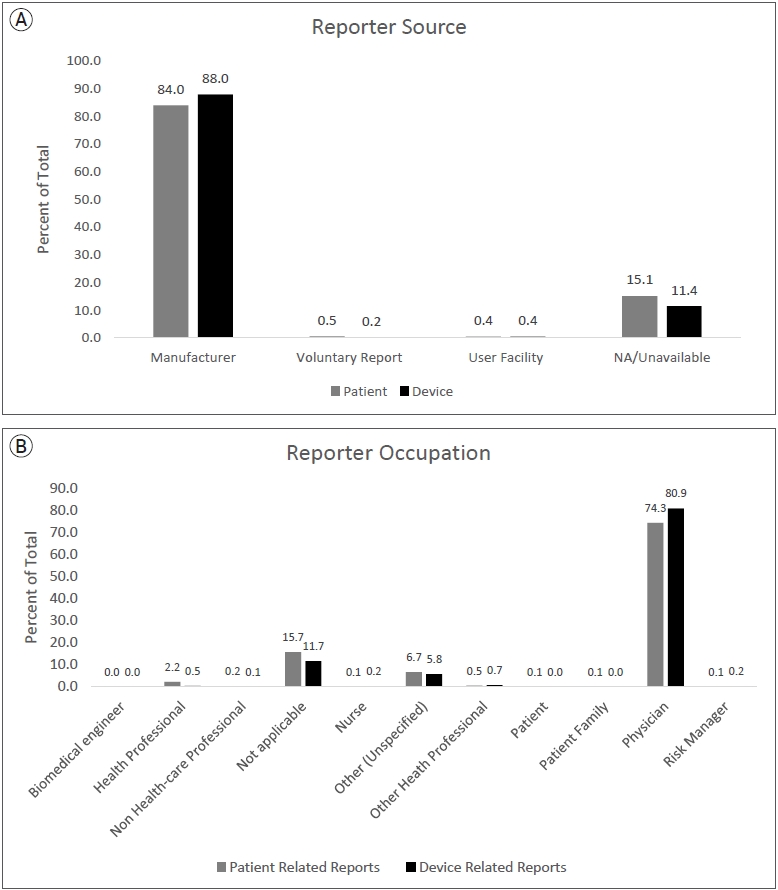
All patient and device related (PR and DR) reports from January 1, 2011, to June 30, 2021, were downloaded and compiled from the MAUDE database. Since MAUDE data is updated as new information is received, this data is current as of the extraction date on February 5, 2022. Using the variable “DEVICE_REPORT_PRODUCT_CODE”, reports relating to flow diverter stents were extracted using the device identifier code “OUT”, and those pertaining to devices other than pipeline were excluded. Using the medical device report (MDR) code which is consistent across all the raw MAUDE data files, PR and DR data were matched with the corresponding MDRs. These reports were further filtered into those representing PR and DR events. The data selection and filtering process is outlined in Fig. 1. Fig. 2A shows the source of reports and Fig. 2B shows the reporter occupations.

Flowchart of the data capturing and filtering process.
Report origination.
Raw PR adverse events were further categorized into broad categories generated by the authors to group similar adverse event reports. Broad author created groups for PR adverse events were Cerebrovascular, Death, Neurological, Decreased Therapeutic Response, Somatic Symptoms, Other, Foreign Bodies, Visual Symptoms, Cardiovascular, and Implant Failure. Sorting PR raw reports into these groups was conducted to combine possibly overlapping terms, such as types of intracranial hemorrhages, to provide a broader clinical view. Broad author created groups for DR adverse events were Operational Failure, Mechanical Failure, and Quality Issues. Operational Failure was defined as difficulties in activating, using, or navigating the device not directly related to mechanical features of the device. Mechanical Failure was defined as inherent mechanical issues with the devices. Quality Issues were defined as reports of compatibility or other quality issues that cannot be classified elsewhere. The classification was conducted and reviewed independently by two neurosurgeons. The process and definition of author generated classifications for PR and DR related reports are shown in Supplementary Fig. 1. Frequencies of each PR and DR report were calculated, along with author generated categories. Reports were further classified by reporter occupation. The structure and linkage of content within the MAUDE database has been described in a previous publication [10].
Statistical analysis
Data filtering and organization was conducted using R-Studio version 2021.09. Data in this study were examined using descriptive statistics to evaluate for frequency of reporting. The FDA suggests MAUDE data cannot be used for inferential statistics or to derive trends due to the nature of spontaneous reporting.
RESULTS
Pipeline reports
A total of 4733 medical device reports (MDRs) for pipeline stents were collected during the 126-month study period. Each MDR contains information about all DR and PR events for each reported incident. Within all MDRs for pipeline stents during the study period, there were 3489 DR and 1826 PR adverse events reported. The list of all raw PR reports and author generated categories are shown in Table 1. The most frequently reported PR adverse event categories were cerebrovascular (58%), death (13%), and neurological (8%), decreased therapeutic response (7%), somatic symptoms (6%), other (3%), foreign bodies (2%), visual symptoms (2%), cardiovascular (1%), and failure of implant (1%). Specific raw PR reports were highest for terms related to cerebral infarctions/ischemia (14.8%), thrombi/emboli/occlusions (13.4%), and hemorrhages (12.3%) which encompass a significant portion of cerebrovascular reports shown in Table 1. Most frequently reported DR adverse events categories were operational failure (72%), mechanical failure (27%), and quality problems (0.3%). Specifically, activation failure, classified into operational failure, was the highest reported raw DR related term (42%). Fig. 3A shows the author generated categories of PR adverse events and Fig. 3B shows author generated categories of DR adverse events. All raw DR reports are shown in Supplementary Table 1. For MDRs with an identified reporter occupation, only 8 PR reports and 3 DR reports were sent by non-healthcare workers.

Patient related reports
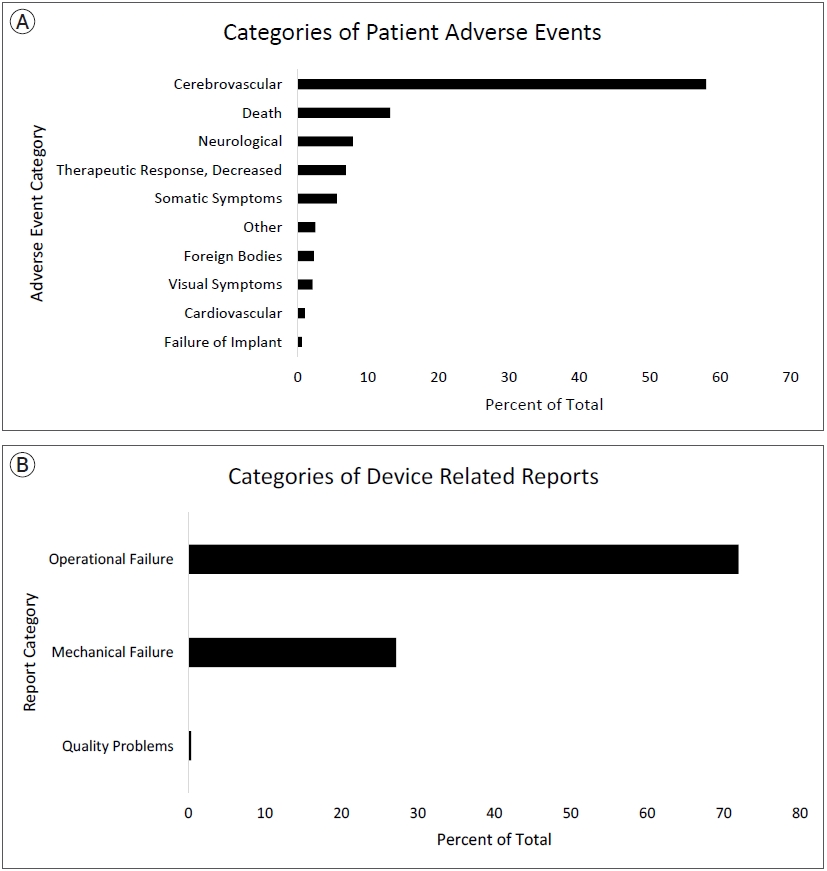
Author generated adverse event categories.
DISCUSSION
FDS are used to treat cerebral aneurysms by diverting blood away from the aneurysm itself to reduce risk of rupture and its subsequent sequela or other neurological deficits, such as cranial nerve deficits related to aneurysm mass effect. Several studies after FDS insertion have been conducted to assess for severe complications such as disability and death [5,22,25]. Rates of overall complications have been reported as high as 17% [28]. However, there are currently no post-market surveillance studies using large databases on more comprehensive patient and device related adverse events. While post-market surveillance for medications is a well-developed methodology for detecting uncommon adverse events [3], medical device surveillance has many unanswered challenges [20]. Some unique challenges compared to pharmacovigilance include device mechanical failures, necessary operator training and learning curve, and lack of established statistical methodology for research studies. With these challenges in mind, analysis of the MAUDE database has previously uncovered significant concerns with medical devices [16]. The characteristics of the MAUDE database that provide a unique perspective compared to previous studies include possessing large scale and longitudinal data maintained by the FDA which has the potential to uncover both rare and common safety issues in the long-term. Although reporting is spontaneous and only mandatory by manufacturers, importers, and device-user facilities, post-market surveillance has been critical in device safety monitoring and encouraging improvement of public safety databases [2,13,17,21].
Our exploration of the FDA MAUDE database for adverse events and device problems related to PEDs revealed that cerebrovascular complications were the most frequently reported adverse event, with cerebral infarction being the most common at a rate of 9%. It is known that the metallic surface of the FDS presents a risk for platelet activation and subsequent thrombus formation even in the setting of dual antiplatelet therapy [9]. Therefore, it is essential to initiate dual antiplatelet therapy in patients with PEDs to reduce the risk of thromboemboli; however, this is conversely associated with an increased risk of hemorrhagic complications [23]. Much literature exists discussing the effects of dual antiplatelet therapy in flow diversion medical management, and our results show that further exploration of these therapies is beneficial to address the most significant complication of FDS. Previous studies report significant cerebrovascular complications associated with FDS with overall rates as high as 10.3% [19]. This post-market surveillance study reinforces that cerebrovascular complications are potentially the leading concerns for adverse events associated with using FDS for intracranial aneurysms. The next highest reported category was neurological complications at a rate of 7.8%. Previous studies report at least one neurological complication after deployment of FDS in up to 18% of patients [5]. However, these studies are often unable to accurately separate primary neurological sequalae from sequalae as a result of cerebrovascular complications, similar to the results presented in this study. The high reports of cerebrovascular and neurological complications are unsurprising, given the patient population and nature of the procedure. Decreased therapeutic response is a broadly defined adverse event within the MAUDE database which likely represents lack of adequate aneurysm obliteration after stent deployment. In our analysis, this adverse event was present in 6.9% of reports. This alludes that failure of aneurysm obliteration is commonly reported but no conclusions can be made regarding the incidence of failure since the total number of pipeline devices deployed is not available in the MAUDE database.
The most frequently reported device problem was operational failure, with activation failure being the most common. Although an inherent limitation of the MAUDE database is a lack of concrete term definitions, we speculate that activation failure refers to the stent being partially or not fully expanded within the patient’s blood vessel or that the stent pusher wire was not able to advance inside the microcatheter. Activation failures and associated device problems may increase the need to install new devices in a telescoping fashion as a rescue strategy given that the PED cannot be retrieved; this also results in an increase in the cost and duration of the procedure overall. Although PEDs are considered cost effective when considering lifetime rehabilitation and treatment costs [26], device problems must still be considered when comparing FDS to other aneurysm therapies given the high device and procedure cost [7]. This represents the first large study to analyze and report device related and quality issues associated with pipeline devices. It is imperative to further research the socioeconomic impact of FDS and alternative therapies given our frequent reports of DR complications.
The vast majority of adverse PR and DR events were sent by manufacturers and reported by physicians as opposed to other healthcare professionals or patients. Essentially, this means that most reports consisted of physicians reporting to the manufacturer or representative, who then compile the information for submission to the FDA. This indicates a crucial lack of direct physician reporting to the MAUDE database. This phenomenon with the MAUDE database has been previously reported, urging the need for increased physician reporting for objective, unbiased data for medical devices [18]. However, several challenges arise including lack of awareness, time-consuming reporting processes, and the relatively unstructured reporting logistics. These findings further emphasize that training and encouraging physicians to self-report adverse events can improve the accuracy of post-market surveillance studies for medical devices moving forward. We also emphasize the importance of developing a comprehensive, transparent, and harmonized post-market surveillance system for medical devices since currently the MAUDE database has several limitations. Firstly, adverse event reporting of devices should encourage reporting and implement additional requirements from hospital administration to obtain a comprehensive dataset. Additionally, the reporting process should include a standardized system to ensure details and categorization of events are unambiguous as currently there are several overlaps and unclear definitions, increasing the difficulty and significance of analysis. Transparency and validity of data is difficult to evaluate due to incomplete disclosure of data [13]. These issues have been noticed in post-market surveillance by the European Union, which has implemented an extensive new system with several requirements to improve the database for future studies [21]. This study further reinforces the need for the FDA to follow and implement advancements towards the MAUDE database to solidify future research for medical devices.
Limitations
The use of the FDA MAUDE database has several limitations. Health care providers are not individually required to report adverse events and thus do not report them for many reasons. Because this database is based on self-reporting, many providers do not report due to time constraints, difficulty in reporting, and lack of knowledge of the submission process or the existence of the MAUDE database [14,18]. Therefore, the majority of reports originate from manufacturers, increasing the potential for bias. Various sources may report the same complications using different descriptions, and thus duplicate reports may be discounted. Additionally, the FDA suggests MAUDE data cannot be used for inferential statistics or to derive trends due to the nature of spontaneous reporting. The data represented in this study reflect a descriptive analysis of reports. Since the MAUDE database does not report the total number of devices deployed, and mandatory reporting of all adverse events is not required, statements of adverse event incidence cannot be drawn from this study. It is important to note that cerebrovascular and neurological complications may overlap in definitions in this study. For example, paresis, classified as a neurological complication could be the result of stroke which is classified as cerebrovascular. Due to the nature of the database and the reporting process, it is impossible to determine cause and effect of the various reported terms. However, each report classified in author generated categories represents a unique report within the database. Further studies are imperative to delineate these categories due to these inherent limitations of the MAUDE database. Despite these limitations, our study reveals the potential adverse events and device issue associated with FDS use, reported in the largest post-market surveillance device database. This may be used towards device development and patient counselling. We also emphasize the need for improvement of the MAUDE reporting system to increase the accuracy and strength of future studies, as post-market surveillance of devices is challenging in its current state.
CONCLUSIONS
Flow diverting stents are a groundbreaking innovation in the treatment of intracranial aneurysms; however, post-market surveillance of adverse events of FDS is limited. Post-market surveillance is important to guide patient counselling and identify adverse events and device problems that were not identified in initial trials. We describe the first post-market surveillance study of reported adverse events with PEDs within a large, public database. Although there are inherent limitations to the MAUDE database, our results highlight some important PR and DR complications that can help optimize patient counseling and management for patients with intracranial aneurysms. Furthermore, we urge optimization of the MAUDE reporting process and to increase awareness to promote objective, direct reports by healthcare workers for accurate post-market medical device studies.
Supplementary figure and table
PR event data classification process
Specific device related reports
Notes
Disclosure
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.