Systemic macrophage depletion attenuates infarct size in an experimental mouse model of stroke
Article information

Abstract
Objective
Macrophages have been shown to play important roles in various pathophysiological processes of the central nervous system via neuroinflammation, leading to an increased interest in macrophage biology. Circulating blood monocytes are among the first cells to infiltrate the brain after ischemic stroke; however, the role of innate immune cells such as monocytes and macrophages remains to be elucidated. Here, we investigated the association between blood monocytes and infarct size following ischemic stroke.
Methods
We induced stroke using a focal ischemia mouse model through middle cerebral artery suture occlusion. To deplete circulating blood monocytes, clodronate was injected intraperitoneally 24 h before the surgery. Animals were sacrificed at specified time points, and the infarct size and mRNA expression were then measured.
Results
The clodronate-injected mice showed significantly smaller infarct size than the control mice. Immunohistochemical staining revealed that monocyte depletion significantly blocked the infiltration of macrophages and microglia. The mRNA expression levels of macrophage and microglia markers were higher in the left infarcted brain than in the right non-infarcted brain.
Conclusions
In summary, monocyte depletion reduced the infarct size and mitigated neurological deficits in mice following ischemic stroke, likely by blocking the infiltration of inflammatory cells such as macrophages and microglia.
INTRODUCTION
Ischemic stroke is a leading cause of disability and death worldwide, resulting in significant socioeconomic and clinical impacts [3]. While the intensive search for new ischemic stroke treatments continues, the current methods have shown limited effectiveness owing to the complex mechanisms involved in the pathogenesis [11]. Recent studies on post-stroke inflammation showed a bidirectional interaction between the brain and immune system after ischemic stroke-induced immunosuppression [11]. In addition, peripheral immune cells have been shown to migrate to the ischemic brain and contribute to neural injury after ischemic stroke [2]. Recent studies reported both the beneficial and detrimental effects of the innate immune system and established that the timing after ischemic stroke is a key factor in determining whether immune responses and inflammation are neuroprotective or neurotoxic. However, the exact role of the innate immune system has not yet been fully elucidated. Although a previous study demonstrated that monocytes/macrophages prevented the transformation to hemorrhagic infarction, the comprehensive contribution of these cells after ischemic stroke remains unclear [5]. Macrophages are phagocytic cells that play two very important roles: initiate the innate immune response and direct the acquired immune response. Macrophages are activated by interferon gamma (IFNγ) or lipopolysaccharide, and they function to eliminate pathogens and mount inflammatory responses [7]. Although it is known that the immune response to ischemic stroke plays a vital role in both the resolution of primary injury as well as the aggravation of secondary injury, the role of infiltrating monocytes has largely been neglected. Therefore, we hypothesized that monocyte depletion in a mouse stroke model would result in a smaller infarct size. To test this hypothesis, we evaluated the consequences of non-selective monocyte/macrophage depletion at the acute phase after the induction of experimental ischemic stroke in mice.
MATERIALS AND METHODS
Experimental animals
All experimental protocols were approved by the Chonnam National University institutional research board (Approval No. TMP-2013-1239). Ten to twelve-week-old male C57BL/6 mice (total=208), weighing 20-25 g, were purchased from Samtako Bio Korea (Osan, Korea) and housed in a pathogen-free barrier animal facility. Animals were maintained on a standard diet and water ad libitum in a temperature-controlled environment under a 12-h light/dark cycle. Every effort was made to minimize the number of animals used and their suffering.
Mouse model of transient focal cerebral ischemia
Anesthesia was induced via the administration of 5% isoflurane and was maintained using 1-2% isoflurane during surgery. The animals’ core body temperature was monitored using a rectal probe and was maintained at 37±0.5°C using a surface heating pad during surgery. Focal ischemia was generated as previously described [13,17]. Briefly, under an operating microscope, the left common carotid artery (CCA) and external carotid artery were exposed through a ventral midline neck incision and ligated proximally. A 6-0 silicon-coated nylon suture with a 0.23-mm tip diameter (Doccol, Redlands, CA, USA) was inserted through the arteriotomy in the CCA just below the carotid bifurcation and advanced approximately 8±0.5 mm until mild resistance was encountered. The suture was then tied with a 5-0 black silk suture at the proximal CCA bifurcation to prevent withdrawal. After 60 min of ischemia, reperfusion was achieved by removing the suture in both the control ischemic and clodronate-injected groups.
The mice were randomly divided into two groups: the control group, which was administered liposome-encapsulated phosphate-buffered saline (PBS) in the lower right side of the abdomen, and the experimental group, which was administered liposome-encapsulated clodronate via intraperitoneal injection to deplete monocytes. Both injections were administered 24 h before ischemic insult. The liposomes were administered at the recommended dose of 10 µL/g (approximately 200 µL) [20,27]. In each group (n=10), all animals were subjected to 60 min of ischemia followed by 3, 5, or 7 days reperfusion and then sacrificed according to the study protocol.
Several groups have previously reported that the administration of liposome-encapsulated clodronate abrogates circulating monocytes in the blood for 3 days [1,10,27]. Since this model of monocyte depletion is well established, we did not perform additional experiments to confirm their depletion in circulating blood.
Infarct size measurement
Infarct size was measured 3 days after the induction of stroke via 2,3,5-triphenyltetrazolium chloride staining (n=10/group, total=20) and was expressed as a percentage of the hemisphere volume, calculated using the following formula: ([contralateral hemispheric volume–ipsilateral non-infarct hemispheric volume]/contralateral hemispheric volume)×100%. Immunochemical staining of resident microglia was conducted using an anti-ionized calcium-binding adapter molecule 1 (Iba-1) antibody to compare the infarct size to the infiltration patterns between the two groups. For Image J analysis, the infarct size in each slice was traced and measured by manually outlining the margins of the non-ischemic areas using Image J 1.80 (NIH, Bethesda, MD, USA).
Immunohistochemical staining
On days 3, 5, or 7 after 60 min of ischemic injury, the brains were removed from mice treated with clodronate or PBS (n=10/group, total=60). After standard histological processing, hematoxylin and eosin-stained sections of the brain specimens were reviewed to determine the infarct area. Tissue blocks of the infarct areas were then selected for formalin fixation and paraffin embedding.
Immunostaining was performed using the standard avidin-biotin complex method with some modifications [22,28]. Briefly, experimental brains perfused with 4% Paraformaldehyde fixative and immersed in a 4% aqueous solution of formaldehyde–10 to 30% sucrose solution for 12 h by each step. Tissue blocks embedded in paraffin and representative paraffin blocks were sliced into consecutive 4-μm-thick sections, and immunohistochemical staining was carried out using a Sequenza staining rack (Thermo Shandon, UK). The sections were deparaffinized in xylene and treated with 0.3% H2O2 in methanol for 20 min to block endogenous peroxidase activity. The sections were then heated in a pressure cooker containing 10 mM citrate-phosphate buffer (pH 6.0) for antigen retrieval. Next, the sections were incubated with anti-Iba-1 antibody (1:400, Abcam) at 4°C overnight. The streptavidin-horseradish peroxidase (Vector Laboratories, Burlingame, CA, USA) detection system was applied to the capillary channels and incubated at 37°C for 1 h. For the chromogen reaction, the tissue sections were incubated with 0.02% diaminobenzidine. The sections were counterstained with hematoxylin and mounted in Universal Mount (Research Genetics, AL, USA). Image J software was used for quantify those section.
Quantification of gene expression via real-time reverse transcriptase polymerase chain reaction (qPCR)
On days 1, 3, 5, or 7 after the induction of ischemic injury, the brains were removed from the mice injected with clodronate or PBS control (n=10/group, total=80). Removed brain was moved CytoVistaTM 2 mm Coronal Mouse Brain Slicer (ThermoFisher Scientific, CA, USA) and sliced with equal thickness for mRNA sample preparation. Representative infarcted area was selected and RNA was extracted from the sliced mouse brain tissues using the PureLink RNA Mini Kit (Invitrogen, Valencia, CA, USA) that included an on-column DNase treatment, according to the manufacturer’s instructions. The RNA concentration and purity were evaluated using a TECAN m200 Pro Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) by measuring the absorbance at 260 and 280 nm to determine the A260/A280 ratio, and a ratio greater than 1.9 was considered highly purified RNA. Extracted RNA (1 µg) was reverse transcribed into complementary DNA (cDNA) using a GeneAmp PCR System 2700, reverse transcriptase, and oligo d(T)20 primers according to the manufacturer’s protocol (Invitrogen). Quantitative real time PCR was conducted to quantify the mRNA expression of chemokine ligand 2/monocyte chemoattractant protein 1 (CCL2/MCP-1) (forward primer: 5′-CAA GAT CCC AAT GAG TAG GC-3′ and reverse primer: 5′-GAT CTC ATT TGG TTC CGA TCC-3′) and integrin subunit alpha M (ITGAM) (forward primer: 5′-ATG CTG CGA AGA TCC TAG TTG T-3′ and reverse primer: 5′-CAG CTG GCT TAG ATG CGA TG-3′) (Applied Biosystems). All reactions were performed in duplicate.
The widely accepted comparative Ct (threshold cycle) method was used to quantify the qPCR results. The arithmetic formula (fold difference=2-ΔΔCt) was used to compare the relative mRNA expression levels in the treated and untreated control mice. Threshold amplification cycle-number data from multiple plates were combined using AB Relative Quantitation software (SDS1.2, Applied Biosystems, Foster City, CA, USA) and the ΔΔCt method.
Statistical methods
Data are expressed as the mean±standard error of the mean and were analyzed using GraphPad Prism software (GraphPad Software 9, San Diego, CA, USA). Statistical analysis of the infarct volume was performed using analysis of variance followed by Newman-Keuls post hoc or Dunnett’s test. A p value less than 0.05 was considered statistically significant.
RESULTS
Out of 208 mice that underwent surgery, 48 died during the observation period (10/38=PBS/clodronate group, mortality rate: 23%). Of the 160 survivors, 80 were randomly assigned to each group (n=10/group), with or without clodronate injection, to measure the infarct size, and the remaining 80 mice were used for quantitative real-time PCR. An equal number of mice was maintained in each group to obtain statistical significance.
Macrophage depletion reduced the infarct size
First, we investigated whether macrophage depletion affected the infarct size after stroke, since it is known that a large number of macrophages can infiltrate the brain and aggravate neuroinflammation. To evaluate the short-term effects of monocyte/macrophage depletion in acute ischemic stroke, clodronate liposomes were administered 24 h before transient middle cerebral artery (MCA) occlusion. Reperfusion was maintained for 3 days by removing the suture from the CCA (Fig. 1A). As expected, the control mice had a mean infarct size of 54.73±3.21% in the ischemic hemisphere, which was significantly reduced to 26.25±2.07% (p=0.006) in the clodronate-injected mice (Fig. 1B, C).
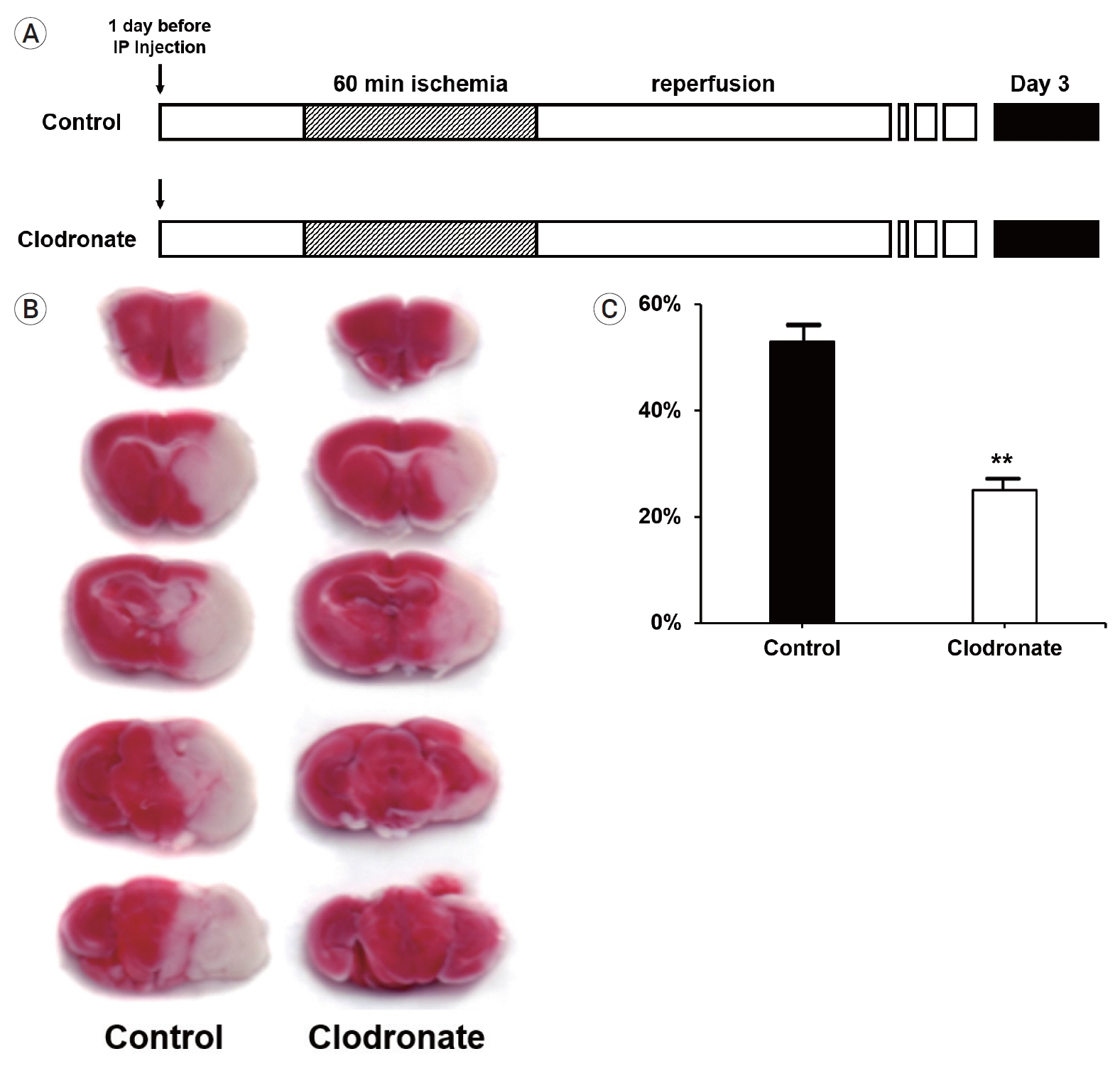
Experimental protocols, 2,3,5-triphenyltetrazolium chloride (TTC) staining, and infarct volume. (A) Both liposome-encapsulated phosphate-buffered saline and liposome-encapsulated clodronate were administered via intraperitoneal injection 24 h before the induction of stroke. After 24 h, focal ischemia was induced in the mice via transient middle cerebral artery occlusion for 60 min, followed by immediate reperfusion for 3 days. The mice (n=10 mice/group) were sacrificed on day 3 (black box). (B) TTC staining of infarcts; representative images. (C) Bar graph of the average infarct volume in each group. Comparison of infarct volume between the clodronate-injected and control mice showing that monocyte depletion had significant protective effects against infarction (54.73±3.21% vs. 26.25±2.07%, p=0.006). Data are presented as the mean±SEM. *p<0.05, **p<0.01 compared to the control group (n=10 mice/group). SEM, standard error of the mean.
Macrophage depletion reduced the infarct volume
Next, we examined the infarct volume at 3, 5, and 7 days after the induction of stroke and macrophage depletion. Mice were injected with either PBS or clodronate 24 h before stroke induction. After 24 h, focal ischemia was induced in the mice via transient MCA occlusion for 60 min, followed by immediate reperfusion for 3 days. The mice (n=10 mice/group) were sacrificed on day 3, 5, and 7 after stroke induction (Fig. 2A).
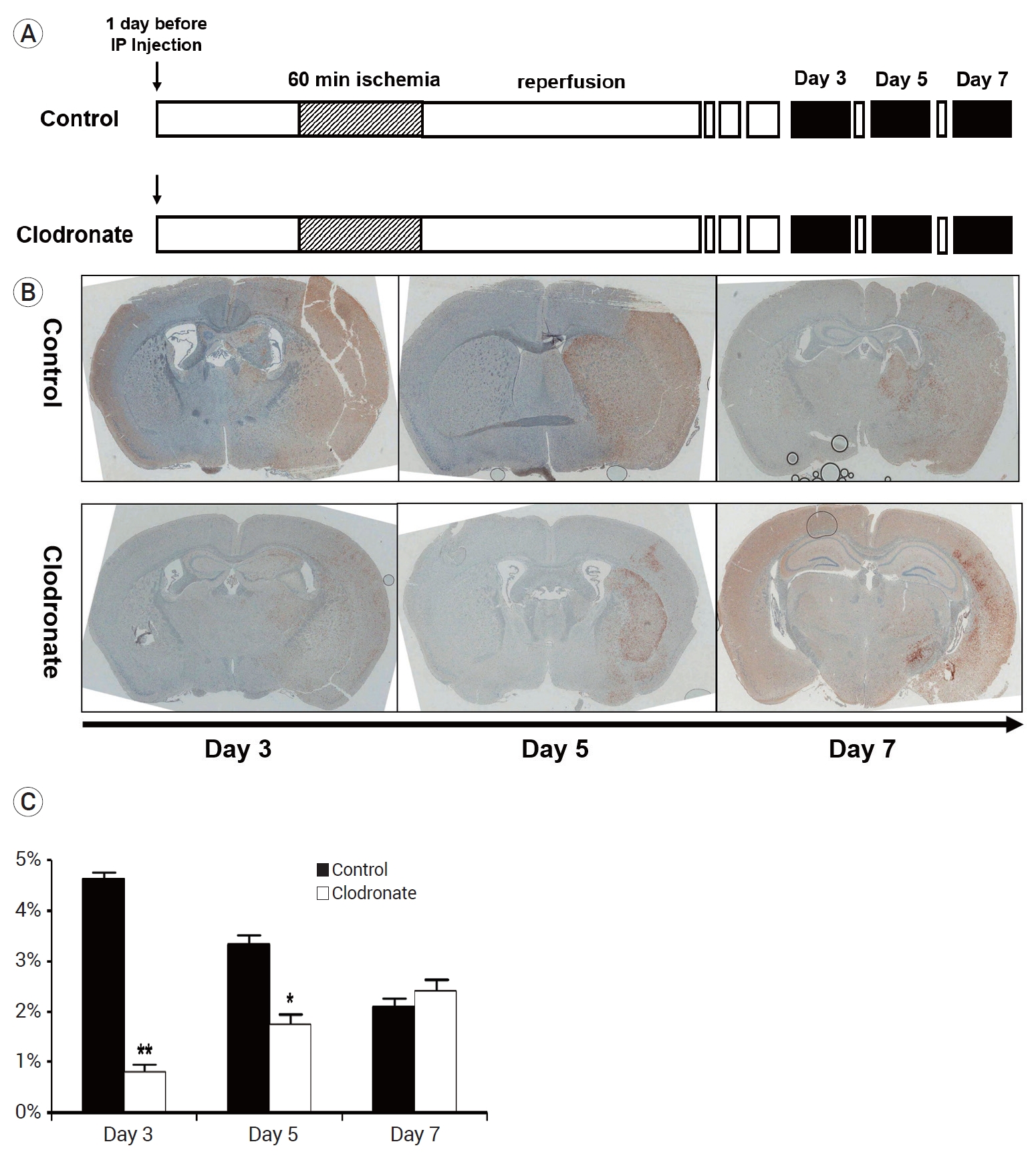
Experimental protocols, ionized calcium-binding adapter molecule 1(Iba-1)-stained cell counts, and infarct volume. (A) The mice were injected with either liposome-encapsulated phosphate-buffered saline (PBS) or liposome-encapsulated clodronate 24 h before the induction of stroke. After 24 h, focal ischemia was induced in the mice via transient middle cerebral artery occlusion for 60 min, followed by immediate reperfusion for 3 days. The mice (n=10 mice/group) were sacrificed on days 3, 5, and 7 after the induction of stroke (black box). (B) Iba-1 staining of infarcts on days 3, 5, and 7; representative images. (C) Cell counts of Iba-1-positive cells in representative infarcts (n=10 mice/group). The clodronate-injected mice showed significantly reduced numbers of resident microglia on days 3 and 5 (4.62±0.2 vs. 0.81±0.22%, p=0.003; 3.33±0.31 vs. 1.74±0.28%, p=0.048; 2.09±0.21% vs. 2.42±0.39%, p=0.76). Data are presented as the mean±SEM. *p<0.05, **p<0.01, and ***p<0.001 compared to the control group. SEM, standard error of the mean.
To evaluate the infarct volume, immunohistochemical staining for Iba-1 (a marker specific for resident microglia) was performed. Our results showed that the number of resident microglia was significantly lower on days 3 and 5 in the clodronate-injected mice than in the PBS liposome-injected control mice (day 3: 4.62±0.2 vs. 0.81±0.22%, p=0.003; day 5: 3.33±0.31 vs. 1.74±0.28%, p=0.048). However, there was no difference between the groups on day 7 (2.09±0.21% vs. 2.42±0.39%, p=0.76) (Fig. 2B, C).
Macrophage depletion affected the mRNA expression of specific markers in both hemispheres
To examine the recovery trends of monocytes/macrophages, we investigated the mRNA expression levels of macrophage and monocyte markers in both the ipsilateral and contralateral brain.
The mice were injected with either PBS or clodronate 24 h before stroke induction. After 24 h, focal ischemia was induced in the mice via transient MCA occlusion for 60 min, followed by immediate reperfusion for 3 days. The mice (n=10 mice/group) were sacrificed on day 1, 3, 5, and 7 after stroke induction (Fig. 3A).

Experimental protocols and mRNA expression in both hemispheres. (A) Mice were injected either with liposome-encapsulated phosphate-buffered saline or liposome-encapsulated clodronate 24 h before the induction of stroke. After 24 h, focal ischemia was induced in the mice via transient middle cerebral artery occlusion for 60 min, followed by immediate reperfusion for 3 days. The mice (n=10 mice/group) were sacrificed on days 1, 3, 5, and 7 after stroke (black box). (B) Integrin subunit alpha M (ITGAM) and chemokine ligand 2 (CCL2) mRNA expression levels in the infarcted left brains on 1, 3, 5, and 7 days are shown. There was no significant difference between the groups on days 1 and 3; however, the control group showed higher ITGAM and CCL2 expression levels on days 5 and 7. (C) ITGAM and CCL2 mRNA expression levels in non-infarcted right brain sections on 1, 3, 5, and 7 days are shown. ITGAM expression levels were consistently decreased after the induction of stroke in the clodronate-injected mice. For days 3-7 after reperfusion, mRNA expression levels in the control and study groups were similar. *p<0.05, **p<0.01, and ***p<0.001. SEM, standard error of the mean.
In the ipsilateral ischemic brain, the mRNA expression levels of ITGAM (a macrophage antigen) in the control group on days 5 and 7 were significantly higher than those in the clodronate-injected mice (day 1: 1.11±0.23 vs. 0.96±0.26, p=0.867; day 3: 1.35±0.09 vs. 1.56±0.07, p=0.912; day 5: 11.70±2.28 vs. 0.8±0.17, p=0.001; and day 7: 41.2±2.31 vs. 0.88±0.33, p=0.001). After day 5, the mRNA expression levels of monocyte chemoattractant protein-1 (MCP-1, encoded by the CCL2 gene) were also significantly higher in the control group than in the clodronate-treated group (day 1: 66.20±6.89 vs. 59.70±6.89, p=0.676; day 3: 92.86±10.52 vs. 60.81±2.03, p=0.691; day 5: 320.40±93.28 vs. 168.21±12.10, p=0.038; day 7: 1767.06±625.68 vs. 330.59±80.15, p=0.001) (Fig. 3B). Simultaneously, in the contralateral brain of the control group, ITGAM expression was significantly higher after day 3 (day 1: 1.03±0.57 vs. 0.94±0.14, p=0.724; day 3: 2.28±0.15 vs. 0.49±0.05, p=0.001; day 5: 1.03±0.03 vs. 0.31±0.04, p=0.001; day 7: 1.08±0.10 vs. 0.29±0.09, p=0.001). As these data show, the ITGAM mRNA expression levels were consistently lower in the clodronateinjected mice. The CCL2 mRNA levels showed a trend to increase over time after stroke induction (day 1: 1.25±0.26 vs. 10.03±0.43, p=0.001; day 3: 4.85±0.07 vs. 10.33±0.98, p=0.001; day 5: 18.25±0.17 vs. 3.99±0.53, p=0.001; day 7: 35.79±0.33 vs. 2.66±0.79, p=0.001) (Fig. 3C).
DISCUSSION
Brain injury after stroke is aggravated by inflammatory reactions, which are regulated by both the innate and adaptive immune systems. The immune response after ischemic stroke plays a critical role in the development and progression of subsequent neuronal death and involves very complex interactions between the peripheral immune system and the resident immune system in the injured brain [11]. Macrophages have been shown to contribute to acute brain injury after stroke. In the brain, macrophages can be derived from either resident microglia [8,18,19] or monocytes that migrate from the peripheral blood [25,26,28]. Resident microglia are the myeloid cells of the central nervous system that are derived from yolk sac progenitors [6]. These cells function as homeostatic regulators in the brain and play a variety of roles after neuronal injury [24]. In response to injury, bone marrow-derived monocytes are recruited to the brain from the peripheral blood [1,16].
Experimental animal studies have shown that the innate and adaptive cellular immune responses to ischemic stroke occur from minutes to a week or even months after injury [4]. After stroke, neutrophils are the first responders; they begin to infiltrate within 30 min of ischemia onset and peak 1-3 days later [33]. Monocytes/macrophages infiltrate from the peripheral blood starting at days 1-2 and peak 3 to 7 days later. In contrast, resident microglia are activated within minutes of ischemia onset and reach maximum levels within 2 days [2]. Thus, local microglia play a role in the early phase of acute ischemic stroke, while circulating monocytes/macrophages likely mediate injury in the later stages [2].
Several studies have demonstrated that monocytes comprise a large percentage of those early infiltrates in the injured brain [2,12,14,28], and the effects of the microglia and monocytes/macrophages present after stroke depend on their M1 (proinflammatory)/M2 (anti-inflammatory) polarization status during disease progression. Impairment of microglial activation was shown to be neuroprotective, suggesting that microglia/macrophages exert a detrimental effect after stroke [9]. At the same time, other studies demonstrated that the inhibition of macrophage infiltration and microglial activation led to unfavorable outcomes such as increased hemorrhagic transformation and worse long-term prognosis, indicating that macrophages and microglial cells have beneficial effects [15,32]. Therefore, a deeper understanding of the transition between the M1 and M2 subtypes in stroke and a better appreciation of the differences between resident microglia and infiltrating monocytes/macrophages are needed to identify potentially therapeutic target cells.
We hypothesized that infiltrating monocytes play a much more important role in the evolution of brain damage after stroke than previously recognized. To elucidate the role of macrophages, it is necessary to examine the effect of depleting macrophages in a complex environment such as an animal model. Systemic macrophage depletion in mice can be achieved via the injection of clodronate-containing liposomes, which promote macrophage “suicide” and have been used in various tissue experiments [31]. Macrophages recognize clodronate-containing liposomes as foreign bodies and engulf them. They are subsequently degraded by lysosomal phospholipases, releasing clodronate and finally inducing apoptosis [30].
Recent studies have shown that clodronate can be injected into different organs using various routes of administration [10,27]. The injection routes can be modified to investigate the role of macrophages in disease pathology [10]. We used the systemic intraperitoneal injection route to intercept the infiltration of circulating blood monocytes, since macrophages in the brain can be derived either from resident microglia or from circulating monocytes.
In our study, we compared the infarct size after ischemic stroke (i.e., at 3, 5, and 7 days after stroke) between macrophage-depleted and control mice using high resolution images. We performed immunohistochemistry using a microglial-specific anti-Iba-1 antibody, since the efficacy of macrophage depletion in a target tissue must be confirmed experimentally and validated either by immunohistochemistry or flow cytometry. We observed some discrepancies in our results of the infarcted brain at 7 days after stroke. We believe that this phenomenon might be due to the recovering late phase of the infarcted mouse brain after day 5.
Chemokine molecules function as inflammatory mediators and seem to play important roles in the early stages of stroke. The overexpression of CCL2 aggravates brain damage by increasing immune cell recruitment and infiltration in the ischemic brain [23]. In addition, it has been reported that antibodies against CXCL8/CCL2 improve stroke outcomes by reducing brain edema and blood brain barrier permeability [21]. Our study also demonstrated increased levels of CCL2 in the control ischemic stroke mice 5 days after the induction of stroke (Fig. 3B). However, very interestingly, we observed discrepancies between ITGAM and CCL2 expression on days 1 and 3 in the control group of the non-infarcted brain (right brain). It is possible that the systemic circulating blood may affect these results in the healthy brain. Further experimental studies are needed to validate this phenomenon.
CONCLUSIONS
Monocyte depletion attenuated infarct size and mitigated neurological deficits in ischemic stroke model mice, likely by blocking the infiltration of inflammatory cells such as macrophages and microglia. Our data suggest pre-injury depletion of monocytes as a novel therapeutic strategy for the treatment of ischemic stroke that has the potential to be translated to human clinical studies.
Notes
Disclosure
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.